Cystic Fibrosis and CFTR
The cystic fibrosis transmembrane conductance regulator (CFTR) is a membrane glycoprotein that is mutated in the autosomal recessive disease cystic fibrosis (CF).
CF is characterized by abnormal ion transport across epithelia. A loss of CFTR functional expression leads to viscous secretions in the airways, pancreatic ducts, and intestine, however many aspects of its pathogenesis remain poorly understood.
CFTR consists of 1480 amino acids and has two membrane domains (TMD1 and TMD2) and two nucleotide binding domains that place it in a large superfamily of “ATP binding cassette” (ABC) proteins.
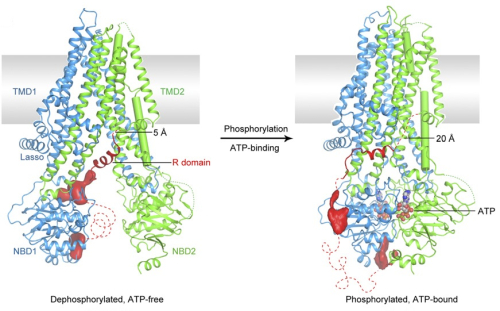
Two functional states of CFTR from cryo-EM
From: Zhang Z, Liu F, Chen J 2018.
Molecular structure of the ATP-bound, phosphorylated human CFTR.
PNAS USA, Dec 11;115(50):12757-12762. doi: 10.1073/pnas.1815287115.
CFTR is the only known ion channel in the superfamily and the only family member that has a regulatory (R) domain. CFTR conducts monovalent anions, the two most important of which are Cl- and HCO3-. It is phosphorylated and activated by cAMP-dependent protein kinase (PKA) and several other protein kinases in the presence of MgATP and dephosphorylated by protein phosphatases 2C and 2A. Phosphorylated CFTR channels open when two ATP molecules are bound at the interface between the nucleotide binding domains and hydrolysis of the ATP at NBD2 enables the channel to close.
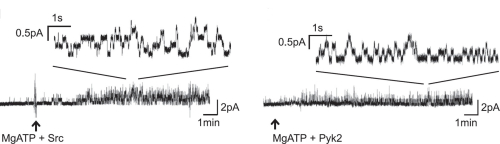
CFTR activation by tyrosine kinases
From: Billet A, Jia Y, Jensen T, Riordan JR, Hanrahan JW 2015.
FASEB J. Sep;29(9):3945-53. doi: 10.1096/fj.15-273151.